

Biology's Longitude Problem
Biotech is the only trillion dollar industry that still feels oddly similar to pre-chronometer Renaissance navigation. We have beautiful instruments and brilliant crews, yet an alarming amount of educated guessing. When I came from engineering into biology, I didn't expect so much of the workflow to boil down to "squint and hope we're roughly here."
Long before GPS, sailors could measure "latitude" easily. A quick look at the sun or a star and they knew their north-south position with decent confidence. But "longitude" was a different story. There was no reliable way to know how far east or west you were.
This gap is obvious in early world maps (e.g. from the Martellus Map (Figure 1) to numerous 15th–16th century charts) where coastlines are distorted, continents are misplaced, and many features were copied from "prior art" rather than empirical measurement. Sailors could follow a stable latitude across an ocean, which made long-distance voyages to India feasible centuries before modern instrumentation, but because no one could determine their actual east-west position, chronic drift was inevitable. It worked mostly well enough though… until it didn't. The 1707 Scilly disaster made this painfully clear: a British fleet misjudged longitude and wrecked four ships, killing ~2,000 sailors.
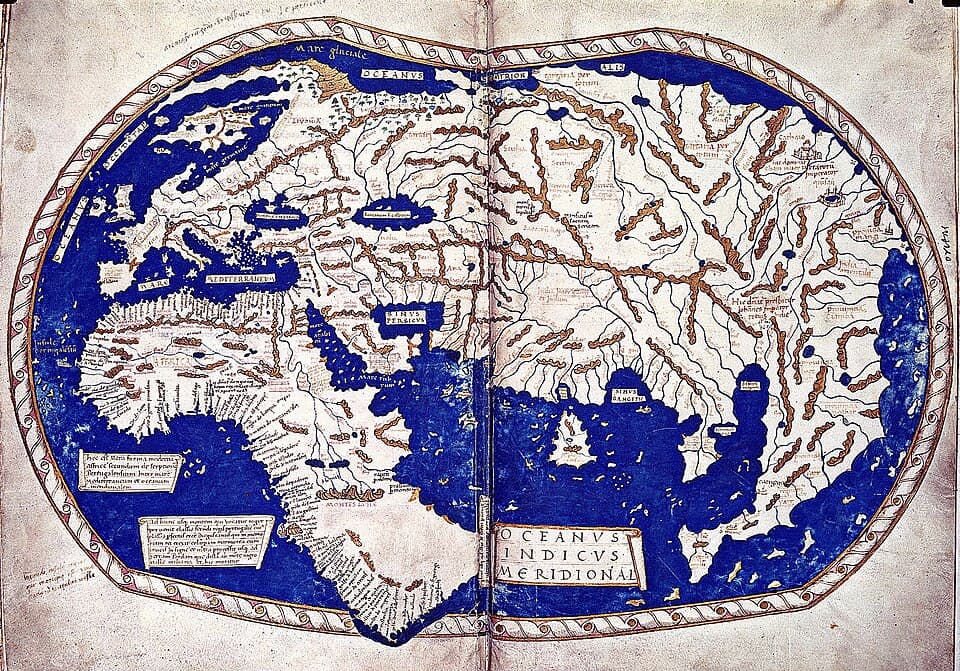
Source: Beinecke Rare Book & Manuscript Library, Yale.
For a long time, the prevailing belief was that better ships, improved seamanship, and more detailed coastal observations would solve these discrepancies. But none of these touched the core constraint: without a stable reference clock, longitude could not be computed. Finally, once John Harrison built a marine chronometer stable enough to keep accurate time at sea, longitude became measurable. And that was when navigation really became "engineerable" and now industries were built on top of it, world maps were redrawn, and trade exploded.
Modern biology feels uncomfortably close to that pre-chronometer period. We have extraordinary instrumentation, yet most discovery programs still depend on indirect proxies, partial readouts, and one-dimensional assays that approximate what is happening inside cells. Better instruments do not fix the missing coordinate. One receptor. One construct. One plate. Some screens take five or six months and produce datasets so specific to a single question that the result cannot describe the wider interaction space.
Ref.
Coming from engineering, this part of biology always surprised me. Most biological measurements sit two or three steps away from the event we actually care about. We perturb a cell, edit a sequence, express a construct, and then measure a fluorescent reporter or proxy phenotype. Want to know if a receptor bound its ligand? We often measure fluorescence downstream of transcription, signaling, and binding. These tools give us estimates, not coordinates.
In large-scale induced Pluripotent Stem Cell (iPSC) engineering projects I worked on, we occasionally saw differentiation drift caused by a weak, unintended interaction. For example, an engineered transcription factor transiently binding a partner outside the target pathway and nudging the timing of a fate decision.
A critique that captured this gap long before CRISPR or AlphaFold existed is Yuri Lazebnik's 2002 essay "Can a Biologist Fix a Radio?" He imagined handing a broken radio to a team of biologists and watching them diagnose it exactly the way we approach cellular systems: disable one component and see which phenotype disappears, overexpress another part to check whether the output increases, and interpret fluctuations in a proxy as evidence of causality. Anyone in biotech who has conducted knockout screens or used pathway reporters has lived some version of this problem. This is how we investigate complex systems when we do not have a wiring diagram.
The missing part is understanding the interactions that govern the system. Biological data is collected as isolated observations rather than as connected systems. We quantify individual proteins, variants, and perturbations while nearly everything we care about emerges from interactions. This core wiring of cellular behavior remains largely unmapped because traditional methods are slow, expensive, and one-dimensional. The value is not just in knowing each interaction. It is in measuring patterns across families, motifs, cross-reactivities, competition groups, and non-binders. Once interactions are measurable, models can learn from a map grounded in quantitative signal instead of navigating from sparse positives.
At some point this stopped feeling like an abstract curiosity and started feeling like a practical engineering problem. If so much of discovery hinges on interactions we can't see, then maybe the solution isn't to build bigger screens or more elaborate reporters, but to measure the interactions directly and at scale, effectively constructing the missing coordinate.
The missing coordinate is an engineering-grade interaction axis built from directly measuring who interacts with whom at proteome scale. And the only way to get it is with a platform that can collide thousands of receptors, variants, and ligands in the same experiment and read out the full pattern of specificity, off-targets, cooperativity, and emergent behavior in one shot. That idea eventually became the motivation behind Lagomics. We're still early, still building, and still discovering the edges of the map. But the goal is to find the missing reference frame so that biologists, computational teams, and drug discovery programs can spend less time guessing and more time reasoning from something solid.
Biology will always surprise us, but it shouldn't do so for reasons we simply forgot to measure. Once interaction data becomes observable at scale, the field can move from reverse-engineering phenotypes to genuinely programmable biology. And when the interaction layer is known, in silico simulation becomes accurate enough that most design-build-test cycles no longer require a full-on multiyear wet lab pipeline in the loop; the lab becomes a validation step rather than the engine of discovery. At that point you don't need billion-dollar programs to produce billion-dollar outcomes, you just need the coordinate we've been missing.