

Why Drug Ads Sound Like Horror Stories
What late-stage side effects reveal about interaction blind spots in antibody drug development
If you live in the U.S., you've seen these ads. A smiling family. Bright colors. Children running through a park. And over it, a calm voice listing serious infections, immune reactions, organ damage, cancer risk, even death. What's easy to forget is that these side effects aren't random. They're the end result of biological interactions that were unknown, underestimated, or only partially understood when that drug was designed. This pattern shows up especially clearly in one of the most successful drug classes of the last few decades: antibody therapies.
Antibodies are highly specialized proteins the body naturally produces, acting like a molecular "lock-and-key" system to precisely find and bind to specific foreign or harmful molecules (like viruses), flagging them for removal or neutralization. Therapeutic antibodies use this targeting mechanism, but are "engineered" to bind to specific disease components, such as inflammatory signals, tumor growth factors, or immune-suppressing "off switches" in cancer.
Think of antibodies as highly effective and customizable smart drugs. They excel because they latch onto a specific target very strongly, stay in the body long enough to work, and their structure is modular. This powerful combination of features has made them the go-to treatment for major diseases like cancer and autoimmune disorders, with over 100 successful products on the market.
Welcome to the Molecular Jungle
In practice, when an antibody drug is administered, it usually finds its specific target surrounded by many other molecules. It affects the main target but also connects with many other unintended molecules, like a specific key that can also open several unintended locks (off-target proteins). These secondary interactions may be weaker, but they can become biologically relevant when:
- The sheer number of wrong locks amplifies a weak interaction into a strong effect.
- Wrong locks grouped together create a higher collective impact.
- Chronic exposure to the "bad key" causes long-term problems.
In other words, context determines the impact particularly in a real-life biological system. While individual lab tests might not show a clear-cut interaction, the cumulative effect of these tiny, often-missed encounters significantly alters the drug's performance. This complex environment dictates how fast the drug is absorbed, where it builds up in the body, how long it stays active, and even how the immune system reacts.
Take Belantamab mafodotin, an approved drug from 2020 to fight multiple myeloma. It caused an unexpected side effect when a high number of patients developed eye problems, specifically corneal damage, severe enough that doctors had to pause treatment and put a special risk-management plan in place.
Even highly specific, targeted drugs, like Antibody-Drug Conjugates (ADCs) and T-cell engagers (a type of bispecific antibody), have frequently hit unexpected walls in human clinical trials. For ADCs, many programs were stopped late in development because the drug became too toxic for the whole body or specific tissues at the needed therapeutic dose, even when the drug successfully hit its intended biological target. Similarly, T-cell engagers, which are engineered for high precision, still led to serious, emergent side effects like cytokine release syndrome and neurotoxicity that only became apparent after the first human trials.
So, how do antibody teams actually manage off-target risk today?
There is no single point in an antibody program where a team definitively "checks for off-targets." Instead, risk is assessed gradually, across different functions, over multiple years, with each stage optimized for a different decision pressure (Figure 1). Antibody programs make their most flexible decisions early, when interaction data is sparse and simplified. Richer biological signals, including off-target effects, often emerge later, after manufacturing, study design, and capital commitments constrain redesign. This timing mismatch explains why safety liabilities are frequently detected when they are hardest to address.
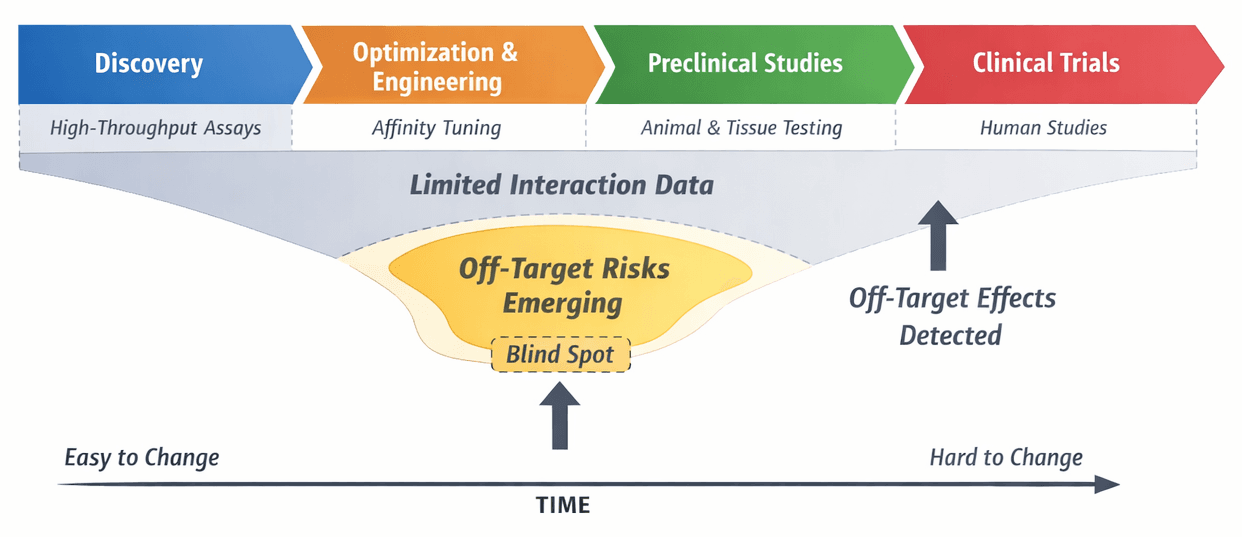
The Slow March Toward Irreversibility
Early discovery (~months 0–12)
The process of finding a new antibody drug starts with thousands of initial candidates, created in labs using various advanced techniques.
The first question to answer is: Does the drug candidate stick to the intended biological target? This is checked quickly using simple, high-volume lab tests. Here, we only look for the intended binding and a very narrow range of unintended binding, usually against very similar molecules or known troublemakers. Proteome-scale screening platforms, such as membrane protein arrays (e.g. Charles River's Retrogenix®)[1], have expanded early specificity assessment across thousands of human targets. While these tools increase breadth, they still tend to produce isolated interaction hits without competitive context or prioritization, limiting their impact on early program decisions.
At the same time, the teams run "manufacturability" checks to filter out molecules that are inherently unstable or difficult to produce at scale (e.g., they clump together or stick to everything). These tests are great for flagging manufacturing problems but aren't designed to predict complex, subtle side effects in the body.
In this early phase, the goal isn't to perfectly eliminate all risks, but to rapidly shrink the massive starting pool of candidates down to a small, manageable set that is worth the huge investment for deeper research.
Optimization and engineering (~months 12–24)
Once the initial promising antibodies are selected, they undergo "humanization," fine-tuning for better binding, and optimization for efficient production, stability, and delivery. These changes intentionally modify the antibody's structure and surface properties. The problem is that these alterations can unexpectedly change how the antibody interacts with other targets, in ways that are tough to predict from earlier testing.
While security checks (for immediate issues) are run again, the intense investigation into unexpected side effects rarely expands. New bugs introduced during this optimization phase often slip by unless they cause obvious, immediate failures.
In this stage, the R&D team's focus has changed. They have started explicitly optimizing for four main metrics, much like balancing features in a new product: Efficacy (how well the drug works), Scalability (how easy it is to mass-produce), Delivery (how effectively it reaches the intended target), and Risk Tolerance (the acceptable level of risk).
Preclinical, tissue and animal studies (~months 24–36)
As drug candidates get closer to human trials, tests are run to see where the drug sticks in different human and animal tissues. These tests satisfy regulators by showing the drug's distribution, but they don't explain what the drug is binding to or if that binding matters biologically.
Next, we move to expensive, slow, and resource-heavy animal studies. These only show the final outcomes (e.g. toxicity or unexpected distribution) not the molecular-level causes behind them.
By this late stage, a massive amount of time and money is already invested in the pharma program. Fixing a problem means a costly and time-consuming redesign. The focus shifts from truly understanding the drug's mechanism to simply deciding if the observed risk is low enough to proceed.
Now Everyone Is in the Room
By the time the question of deeper off-target analysis arises, the program is already advanced enough that the answer carries real cost. Imagine R&D teams (Discovery) have proof that a new drug (target engagement) is working well in early tests, but Safety teams are worried about unexpected side effects (off-target signals) that aren't fully explained yet. The teams handling the development process (Development) have to balance these risks against deadlines, consistency, and the cost of changing the drug's design. Ultimately, leadership has to decide if the remaining risk is worth the money and time already invested.
Adding to this is the non-scientific pressure from investors and partners who are expecting this drug to move forward. Bringing up a list of unresolved side effects at this late stage causes headaches about things like the right dosage, manufacturing costs, study setup, and timelines, and it invites extra scrutiny from regulators. By the time all these pressures build up, the project is usually too far along to easily fix the underlying problem without a major, costly overhaul. A full pivot is often required.
What's actually breaking down here is the interface between interaction-level information and program-level decisions. Early in a program, when experiments are fast and flexible, interaction data is sparse and deliberately simplified. Later, when richer biological signals finally appear, the program is already constrained by manufacturing choices, study design, timelines, and capital. Off-target risk tends to show up precisely in that narrow window, when it first becomes visible at the organism level, but is already difficult to trace back and correct at the molecular level.
So Why Not Just Screen Everything Upfront?
In most antibody programs, early experiments are intentionally narrow because broader interrogation is slow, expensive, and difficult to interpret with today's tools. Expanding scope too early often produces more ambiguity than clarity, for a few structural reasons:
- Pairwise assays strip away context. Standard off-target screens surface isolated interactions outside of any competitive or biological setting, producing lists of weak positives rather than actionable insight.
- Signals arrive without relevance. When a marginal binder appears on an off-target panel, there is often no information about abundance, competition, or tissue specificity. The data cannot indicate whether the interaction will ever matter in vivo.
- Results lack hierarchy. Off-target screening typically returns a list, not a ranked landscape. Everything looks potentially concerning, which in a timeline-driven organization means nothing is acted on decisively.
As a result, programs default to simpler questions that can be answered quickly, which is why "just screening more earlier" has never become standard practice, despite widespread awareness of the stakes.
Seeing the Interactions Early
The missing piece is tooling that can resolve interaction behavior early without exploding cost or decision noise. A system that actually helps must do the opposite: resolve interactions under competition, preserve identity at scale, and return a ranked interaction landscape rather than a collection of binary hits. Only then does off-target screening and safety become something you design for, rather than something you negotiate around after the fact.
This is the gap we are exploring at Lagomics. We are building toward an interaction-first measurement framework that makes these behaviors observable earlier, when programs still have the freedom to act.